

Fenilketonüri (PKU) diğer birçok doğumsal metabolik hastalıkta olduğu gibi otozomal çekinik kalıtım gösteren kalıtsal bir metabolik bir hastalıktır. Cinsiyet ayırımı yoktur, erkek ve kız çocuklarda aynı sıklıkta izlenir. İsmini hastaların idrarında saptanan bir maddeden almaktadır.
Hastalık ilk olarak 1934 yılında Asbjörn Fölling (1888-1937) isimli Norveçli bir hekim tarafında zihinsel geri olan sarışın, mavi gözlü iki kardeşte tanımlanmıştır.
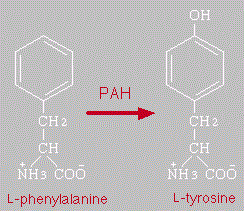
Hasta kişiler proteinli besinlerde bulunan fenilalanin amino asitini tirozin amino asitine çeviremezler. Fenilalanini tirozine çevirmek için gerekli olan ve karaciğerde işlev gören “fenilalanin hidroksilaz (PAH)” enzimi bu hastalarda aktif değildir. Vücutta biriken fenilalanin ve türevleri beyin omurilik sıvısına geçer, burada bileşiklerin düzeyi yükselir ve hasta bireyde zekâ geriliğine, sinir sistemini ilgilendiren birçok bulguya ve nörolojik gelişim geriliğine neden olur. Hayatın ilk birkaç ayında hastalık neredeyse hiç bulgu vermez ve fenilketonüri hastalığı olan bebekleri sağlıklı bebeklerden ayıran özellikler fark edilemez. Zamanında tanı konularak tedavi uygulanmayan fenilketonürili çocuklarda 5-6 aylardan sonra zekâ ve motor gelişimdeki gerilik dikkati çekmeye başlar. Yaşıtlarından farklı olarak destekli desteksiz oturma, emekleme, yürüme, konuşma becerilerini geç kazanırlar ya da kazanamazlar. Hastalık yenidoğan taraması ile saptanıp ilk 3 ayda tedaviye başlanmaz ise hastalığın şiddetine uyan zihinsel özür gelişmesi kaçınılmazdır. Yine tedavi edilmemiş hastaların yaklaşık %60 kadarında ailesine oranla açık saç rengi, açık göz rengi, açık cilt rengi saptanmaktadır. Hastaların vücüt sıvılarında özellikle idrarda özel bir koku dikkati çeker.






{kind=link}